A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19
N Engl J Med 2020;382:1787-99
March 18, 2020
Bin Cao, Yeming Wang, Danning Wen, Wen Liu, Jingli Wang, Guohui Fan, Lianguo Ruan, Bin Song, Yanping Cai, Ming Wei, Xingwang Li, Jiaan Xia, Nanshan Chen, Jie Xiang, Ting Yu, Tao Bai, Xuelei Xie, Li Zhang, Caihong Li, Ye Yuan, Hua Chen, Huadong Li, Hanping Huang, Shengjing Tu, Fengyun Gong, Ying Liu, Yuan Wei, Chongya Dong, Fei Zhou, Xiaoying Gu, Jiuyang Xu, Zhibo Liu, Yi Zhang, Hui Li, Lianhan Shang, Ke Wang, Kunxia Li, Xia Zhou, Xuan Dong, Zhaohui Qu, Sixia Lu, Xujuan Hu, Shunan Ruan, Shanshan Luo, Jing Wu, Lu Peng, Fang Cheng, Lihong Pan, Jun Zou, Chunmin Jia, Juan Wang, Xia Liu, Shuzhen Wang, Xudong Wu, Qin Ge, Jing He, Haiyan Zhan, Fang Qiu, Li Guo, Chaolin Huang, Thomas Jaki, Frederick G Hayden, Peter W Horby, Dingyu Zhang, Chen Wang
PMID: 32187464
DOI: 10.1056/NEJMoa2001282
Chinese Clinical Trial Register: ChiCTR2000029308
Introduction
The new coronavirus (SARS-CoV-2) has infected millions of people and killed at least a million individuals around the world. To decrease the mortality rate of the virus, several potential antivirals have been tested, mainly in patients with severe coronavirus disease-19 (COVID-19). One of the first drugs investigated was that of a human immunodeficiency virus (HIV) type 1 aspartate protease inhibitor, lopinavir, in combination with a complemental drug, ritonavir. These were previously shown to have inhibitory activity against severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV).
Methods
Study design
A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19 was a randomised, controlled, open-label trial in patients with proven severe SARS—CoV-2 infection. The trial was conducted in Jin Yin-Tan Hospital, Wuhan, Hubei Province, China; the epicentre of the pandemic. Understandably but unfortunately, the trial did not involve placebo comparator, since it was designed and conducted early into the epidemic in dangerous and dire circumstances.
Three sources of funding were disclosed; namely, Major Projects of National Science and Technology on New Drug Creation and Development, the Chinese Academy of Medical Sciences (CAMS) Emergency Project of Covid-19, and a National Science Grant for Distinguished Young Scholars. The authors of the study declared no conflict of interests.
The main objective of the trial was to determine the efficacy and safety of lopinavir-ritonavir combination in treatment of severely-ill COVID-19 patients.
Did the trial address a clearly focused issue?
No. Whilst the population studied was randomized properly at the outset and the outcomes were narrowly-defined, an inadequate selection of the population, the lack of placebo, and heterogeneity of the ‘standard care’ received by the patients had seriously compromised the focus of the study.
Eligibility
Inclusion criteria were the following:
- positive reverse-transcriptase–polymerase-chain-reaction (RT-PCR) assay for SARS-CoV-2 in a respiratory tract sample tested by the local Center for Disease Control (CDC) or by a designated diagnostic laboratory.
- Age above 18.
- Pneumonia confirmed by chest imaging
- Oxygen saturation (SaO2) 94% or less on room air or a ratio of the partial pressure of oxygen (PaO2) to the fraction of inspired oxygen (FiO2) at or below 300 mmHg.
Exclusion criteria:
- physician decision that involvement in the trial was not in the patient’s best interest
- presence of any condition that would not allow the protocol to be followed safely
- allergy/ hypersensitivity
- severe liver disease
- drugs contraindicated with lopinavir–ritonavir
- pregnancy or breast-feeding
- known HIV infection
Deviations from eligibility: None have been reported.
Study protocol
357 patients were assessed for eligibility, out of which 199 patients underwent randomzation in a 1:1 ratio. What is important to note is that median number of days from illness onset to randomization was 13 in both groups.
All patients received ‘standard care’. This practically meant giving oxygen, antibiotic treatment, vasopressors, glucocorticoids, renal-replacement therapy, or ECMO when needed. On top of that, the studied half has been receiving 400 mg lopinavir plus 100 mg ritonavir oraly twice daily for 14 days.
Was the assignment of patients to treatments randomised?
Yes.
An outside statistician performed randomization sequence, with web-based allocation concealment system.
Were the groups similar at the start of the trial?
No.
To partially even out an irregular oxygen needs of individual patients, randomization was stratified by the respiratory interventions at the trial’s beginning. However, virology assessment has shown that a total of 69 patients (35%) who had a diagnostic respiratory tract sample that were positive on RT-PCR had a negative RT-PCR result on the throat swab taken after consent. Moreover, viral load at day 1 was higher in the studied group (4.4±2.0 log 10 copies per milliliter vs. 3.7±2.1). This calculation includes only those 130 patients who had positive RT-PCR during trial’s duration, since the rest had undetectable viral RNA levels. Additionally, the groups differed clinically, as well. For example, the number of cancer patients was 5 vs 1 in control group, and there were two more diabetic patients and three more patients with cerebrovascular disease in the control group. Finally, 13 compared to 9 patients in control group were receiving interferon at the beginning of the trial.
Were patients, health workers, and study personnel ‘blind’ to treatment?
No.
This was an open-label trial. This is source of potential bias, as the same clinicians who knew about treatment assignment were analysing the data.
Aside from the experimental intervention, were the groups treated equally?
No.
All patients were receiving individualized ‘standard treatment’ plan. This resulted into notable irregularities in given medication, such as glucocorticoids (32 vs 35 in control group), or vasopressors (17 vs 27 in control group).
Were all of the patients who entered the trial properly accounted for at its conclusion?
Yes.
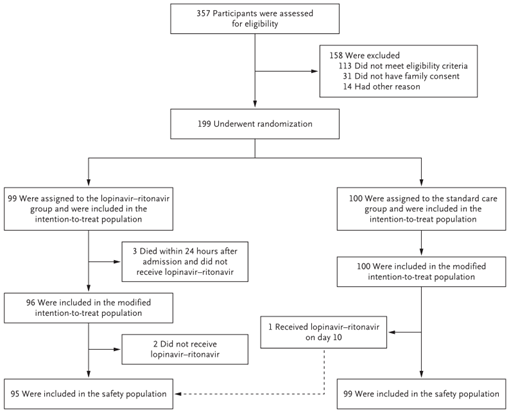
Protocol deviation
Out of 99 in the studied group, 3 died before being given lopinavir-ritonavir. Moreover, 2 patients from study group did not receive lopinavir-ritonavir because the attending physician refused to prescribe them. Finally, 1 patient from the control group received the combination on day 10. All 6 patients were included in the final analysis. Ad hoc analysis was run excluding those 3 early deaths.
Outcomes
The primary end point was the time to clinical improvement, defined as the time from randomization to an improvement of two points (from the status at randomization) on a seven-category ordinal scale or live discharge from the hospital, whichever came first.
Other clinical outcomes included:
- clinical status as assessed with the seven-category ordinal scale on days 7 and 14
- mortality at day 28
- the duration of mechanical ventilation
- the duration of hospitalization in survivors
- the time (in days) from treatment initiation to death
- viral RNA detection over time
- viral RNA titer area under-the-curve (AUC)
- Safety outcomes included
- adverse events that occurred during treatment
- serious adverse events
- premature discontinuation of treatment
Analysis
Primary analysis was on an intention-to-treat basis and included all the patients who underwent randomization. The time to clinical improvement
was portrayed by Kaplan–Meier plot and compared with a log-rank test. Hazard ratios with 95% confidence intervals were calculated by means of the Cox proportional-hazards model. Five above-mentioned patients who did not receive lopinavir–ritonavir group were nevertheless included in the intention-to-treat analysis, since no similar removals occurred in the standard-care group. A modified intention-to-treat analysis that excluded three early deaths was also performed. Post hoc analyses include subgroup analysis for National Early Warning Score 2 (NEWS2) of 5 or below or greater than 5 and those who underwent randomization up to 12 days or more than 12 days after the onset of illness.
In tests for secondary or other outcomes, results are reported as point estimates and 95% confidence intervals. The widths of the confidence intervals have not been adjusted for multiplicity, so the intervals should not be used to infer definitive treatment effects for secondary outcomes.
Results
Primary outcome
How large was the treatment effect? How precise was the estimate of the treatment effect?
At 160 patients, which was the original total sample size, the trial was deemed underpowered. This would provide the trial with 80% power to detect a difference, at a two-sided significance level of α = 0.05, of 8 days in the median time to clinical improvement between the two groups, assuming that the median time in the standard-care group was 20 days and that 75% of the patients would reach clinical improvement. A decision was made to continue enrollment by investigators. In the end, 199 patients were enrolled.
Compared to standard-care alone, the intention-to-treat population did not have a different time to clinical improvement (median, 16 days vs. 16 days; hazard ratio for clinical improvement, 1.31; 95% confidence interval [CI], 0.95 to 1.80; P = 0.09).
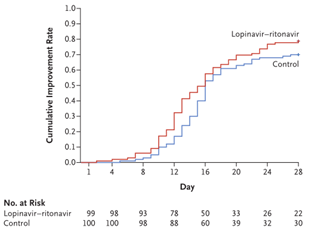
In the modified intention-to-treat population, the data were more promising, but the median time to clinical improvement of 15 compared with 16 days in the standard-care group (hazard ratio, 1.39; 95% CI, 1.00 to 1.91) is still statistically insignificant. Time to clinical improvement was not found to be shorter in the 12 days after the onset of symptoms group (hazard ratio, 1.25; 95% CI, 0.77 to 2.05). No significant differences were observed when the time to clinical improvement was assessed by NEWS2 score.
Secondary outcomes
The 28-day mortality was numerically lower, but was statistically insignificant. These numerical differences were noted both in the lopinavir–ritonavir group in the standard-care group for either the intention-to-treat population (19.2% vs. 25.0%; difference, −5.8 percentage points; 95% CI, −17.3 to 5.7) or the modified intention-to treat population (16.7% vs. 25.0%; difference, −8.3 percentage points; 95% CI, −19.6 to 3.0). Again, lopinavir–ritonavir group had a shorter stay in the intensive care unit (ICU) in absolute numbers than those in the standard-care group (median, 6 days vs. 11 days; difference, −5 days; 95% CI, −9 to 0, but the CI contains 0. A similarly disappointing result was observed in shorter duration from randomization to hospital discharge (median, 12 days vs. 14 days; difference, 1 day; 95% CI, 0 to 3]).
The only statistically significant secondary outcome was the percentage of patients with clinical improvement at day 14 (45.5% vs. 30.0%; difference, 15.5 percentage points; 95% CI, 2.2 to 28.8). This might be coincidental, however, as the mortality in the following days, and indeed the overall mortality at the 28th day, did not show any statistical differences.
There were no significant differences for other outcomes such as duration of oxygen therapy, duration of hospitalization, and time from randomization to death or detectable viral RNA for SARS-CoV-2.
Adverse events
46 patients (48.4%) in the lopinavir–ritonavir group and 49 (49.5%) in the standard-care group reported adverse events between randomization and day 28.
Gastrointestinal adverse events including nausea, vomiting, and diarrhea were more common in lopinavir–ritonavir group than in the standard-care group.
Serious adverse events occurred in 51 patients: 19 events in the lopinavir–ritonavir group and 32 events in the standard-care group.
- There were 4 serious gastrointestinal adverse events in the lopinavir–ritonavir group but none in the standard-care group. These were all attributed to the treatment.
- Respiratory failure, acute kidney injury, and secondary infection were more common in patients receiving standard care.
Discussion
A few antivirals have been proposed for treatment of COVID-19, all with varying degree of evidence. However, only remdesivir is currently recommended in patients with a severe form of the disease via emergency authorization.
In this clinical trial, hospitalized adults with severe COVID-19 have been given the lopinavir-ritonavir combination to figure out the efficacy and safety.
Most importantly, the disclosed data of this trial are not convincing. The study’s design lacked clear divisions between groups, in terms of placebo and supportive treatment. Moreover, the post hoc analyses which tried to account for early deaths further undermined the reliability of these reports.
On the other hand, some of the primary outcomes, whilst statistically insignificant, have shown a small numerical difference, perhaps because of small antiviral effect. This remains speculative, however, and a larger and a more focused trial would be needed to ascertain all this. Similarly, the mortality has shown a feeble numerical difference, without enough statistical power to show a clear benefit.
Adverse effects were noted, but the question of how many of these have been caused by the studied agents and how many by compounding circumstances like the ‘standard care’, the individualized treatment regimes, or actual patient differences remains unanswered or unanswerable.
Can the results be applied to the local population, or in your context?
No. There are limited applications since the peculiarities of the studied groups and the faulty design of the study itself prevents extrapolation to other settings.
Were all clinically important outcomes considered?
Mortality after the 28 days should have been considered. Also, the sequelae and complications of the coronavirus infection, which could take longer to manifest, should have been included. These would perhaps help in determining the effect of studied antivirals and some benefit might have been seen.
Are the benefits worth the harms and costs?
As there are no clear benefits, and there is a rather high percentage of adverse effects, it is unjustifiable to use the proposed combination of antivirals in the treatment of severe COVID-19.
Author and attribution
Tomáš Poliačik is a Junior Clinical Fellow at St Helens and Knowsley Teaching Hospitals NHS Trust. No conflicts of interest have been disclosed.
11 questions from RCT checklist by Critical Appraisal Skills Programme have been used under non-commercial CC 3.0 license.
References
Cao, B. et al. (2020) ‘A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19’, New England Journal of Medicine, 382(19), pp. 1787–1799. doi: 10.1056/NEJMoa2001282.
John Hopkins University. Coronavirus COVID-19 global cases. (2020). Available at: https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6 (Accessed: 22 May 2020).
Baden, L. R. and Rubin, E. J. (2020) ‘Covid-19 — The Search for Effective Therapy’, New England Journal of Medicine, 382(19), pp. 1851–1852. doi: 10.1056/NEJMe2005477.
Chan, J. F.-W. et al. (2015) ‘Treatment With Lopinavir/Ritonavir or Interferon-β1b Improves Outcome of MERS-CoV Infection in a Nonhuman Primate Model of Common Marmoset’, Journal of Infectious Diseases, 212(12), pp. 1904–1913. doi: 10.1093/infdis/jiv392.
Chen F, Chan KH, Jiang Y, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol 2004; 31: 69-75. (no date).
Chu CM, Cheng VC, Hung IF, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax 2004; 59: 252-6 (no date).
Hung, I. F.-N. et al. (2020) ‘Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial’, The Lancet. doi: 10.1016/S0140-6736(20)31042-4.
Sheahan, T. P. et al. (2020) ‘Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV’, Nature Communications, 11(1). doi: 10.1038/s41467-019-13940-6.